QSPRpred App: Predicting Molecular Initiating Event Activation based on chemical structure
Linde Schoenmaker
Introduction
The QSPRpred app is designed to perform hazard identification. It does this by predicting the interaction between a chemical of interest and relevant biological targets (called molecular initiating events (MIEs).
It should be used to perform hazard identification for:
chemical-induced Parkinson’s disease (MIE = metabotropic glutamate receptors)
thyroid-mediated neurodevelopmental toxicity (MIE = thyroid hormone receptors)
MIE activation is an important part of safety assessment on the VHP4Safety platform. It links the presense of a chemical in the human body to a cascade of (known) responses that ends with an adverse outcome (AO).
This prediction is done by target-specific, pretrained QSAR models. QSAR models are machine learning models trained to predict properties of chemicals. These models were trained on curated datasets from the Papyrus database (more information Papyrus). Model predictions are accurate for chemicals similar to the training set. Highly different molecules do not fall within the domain of applicability (an indication of this will be given by the tool).
Accessing the Tool
Access the tool here:
no prerequisites
Tool Functionalities
The app’s initial interface allows users to:
select a MIE model
specify the chemical(s) of interest (Figure 1)
Each model box displays key model characteristics.
A full model description is provided as a QMRF, including:
general information
endpoint (i.e. the biological target of the MIE)
algorithm
domain of applicability
goodness-of-fit
predictivity
interpretation
miscellaneous information
Molecules can be input in two ways:
as comma-separated SMILES strings
via a CSV file containing a “SMILES” column
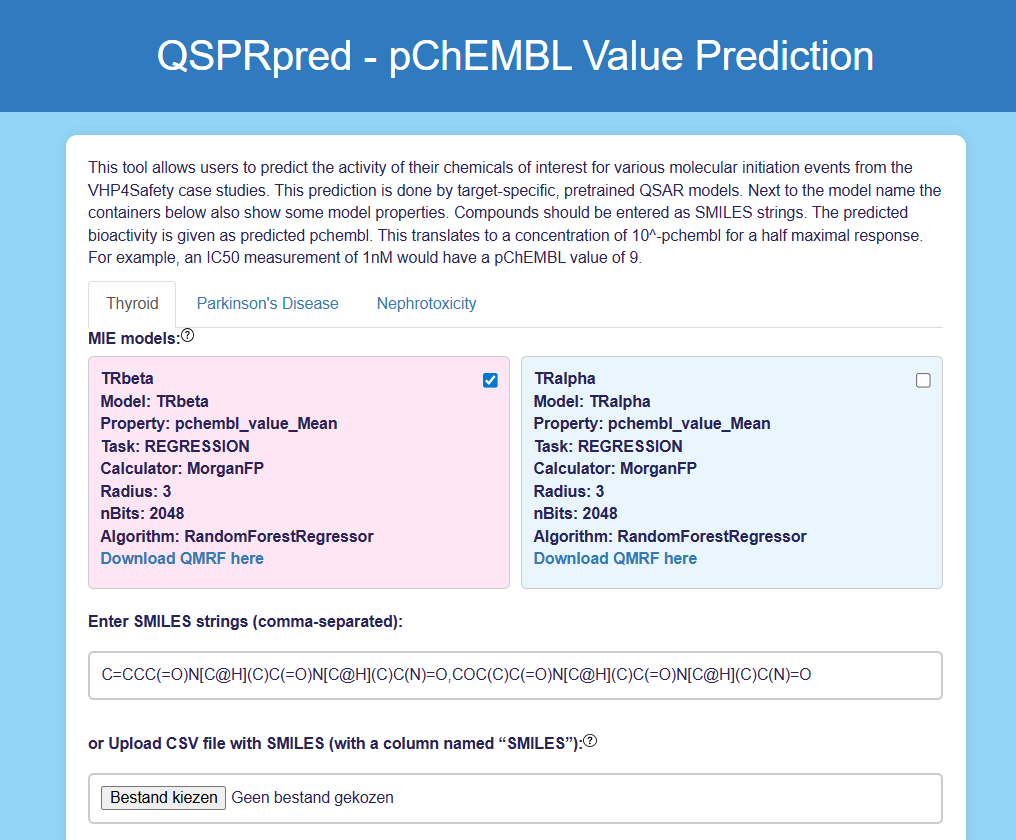 Figure 1: User Interface of the QSPRpred App. The user can select between TRbeta (selected) and TRalpha models and input the desired chemical(s) as SMILES strings.
Figure 1: User Interface of the QSPRpred App. The user can select between TRbeta (selected) and TRalpha models and input the desired chemical(s) as SMILES strings.
Interpreting the Output
Depending on the model the readout is either a numeric value or an activity class (active/inactive). In both cases the endpoint is based on the pChEMBL value (more information pChEMBL). Briefly, this value is a negative logarithmic measure for activity and translates to a concentration of 10^-pchembl for a half maximal response. For example, an IC50 measurement of 1nM would have a pChEMBL value of 9. More information about each model can be found in the QMRF document.
After selecting a model, the user can start the prediction by clicking Run Prediction.
This executes the chosen QSAR model(s) and produces activity predictions.
Along with the prediction value, an indication of the domain of applicability is always shown.
Additional details are available under the Comprehensive output tab, including:
information on the nearest neighbor from the training set
its structure
similarity to the input molecule
measured value
data source(s)
Each prediction is also accompanied by a QPRF document, which includes:
information about the input substance
the model used
the prediction
the input
applicability domain
reliability
(all model-available information is pre-filled, though the document is not fully completed)
The QPRF is intended to support the user by summarizing and documenting key information about QSAR predictions.
For risk assessment, the MIE prediction gives an indication of chemical hazard. When binding surpasses a certain threshold the chemical poses a chemical hazard. Derive a complete safety assessment by combining chemical hazard with internal exposure (derived from toxicokinetic models) and quantitative modelling of the adverse outcome pathway.
Summary
QSPRpred lets users predict MIE activation for their chemical(s) of interest. This is useful for hazard identification of specific chemicals. Within the VHP workflow, MIE activation is used to link internal concentrations to quantitative adverse outcome pathways (qAOPs). Read our preprint for more information about the models for chemical-induced Parkinson’s disease. Explore the tool further using our practice exercises.